Help on using this tool
Skip to interpreting results (below)
Searching for a motif in a set of proteins
If you have a set of sequences that you think might have a motif, then it is simply a matter of
pasting these sequences (in fasta format) into the first box, selecting the species and clicking 'search'.
You don't need sequences, however; you can just input either HUGO gene symbols (e.g. TP53, MDM2), Uniprot
accession (e.g. P04637, Q00987) or identifier (e.g. P53_HUMAN, MDM2_HUMAN). You'll need to tell
the tool whether you are given sequences or accessions in the first box.
If you know, or suspect, that the proteins above have a common interaction partner protein, then you can
specify this protein using either fasta, gene name, accession or identifier (in the second input box). The
tool guesses which input your are giving (i.e. if there is a ">" then it presumes fasta).
Searching for motifs in a set of protein-protein interactions
If you have a set of interactions and suspect these might contain motifs that somehow mediate them, then you can simply paste the interactions into the first box, select the right species, select Interactions as the input type, and click search.
Advanced oiptions
The default options (i.e. those that you see when you load the page) are optimised for a relatively small set of sequences (e.g. 5-10) such that you should find all interesting motifs present. However, there are some situations where you might wish to change them. Below, each parameter is explained, including scenarios when (and how) you would alter them.Sequence filtering
Filtering input sequences is an improtant step in motif hunting for several reasons. We thus have four different means to do this that are detailed below. Sequence homology redundancy filtering: this removes regions that are homologous (assessed by BLAST) to other regions already in the set. If homologous sequences are in a set where motifs are sought, then motifs can be found that arise solely because of descent from a common ancestor, and this will almost always score better than real linear motifs, which generally arise convergently. You can turn this off, in exceptional circumstances, by clicking 'No'.Uniprot region filtering: we have observed, particularly for large sequence sets (e.g. >=50) that it is possible for structural features to give rise to motifs. For instance, with 50+ sequences on can often see patterns like LxxLLxxL that correspond to helices. Leucine is the most common amino acid, so in a set of 50 proteins it is likely that one will find 5 or more sequences that contain such a pattern. We thus also, by default, remove regions annotated in Uniprot as structure features unlikely to contain motifs (specifically: STRAND, HELIX, TRANSMEM, SIGNAL, DISULFID, METAL or COILED). You can turn this off, in exceptional circumstances, by clicking 'No'.
Ordered sequence (IUpred) filtering: Motifs tend to occur in disordered regions (90% or more), so it sometimes makes sense to filter regions that are ordered. However, by default, we do not do this as we found that many real motifs are removed by doing this, which is probably a function of imperfect disorder prediction, or the fact that motif instances are often in short areas of local order in a region that is otherwise disordered. You can, however, turn this filter on (by clicking 'Yes').
Structure domains filtering: As above, it is also possible to define ordered segments more explicitly by matches to known globular domains. For this purpose, we use BLAST to identify sequence similarities to a set of globular domains as defined by the SCOP database. By default, we do not use this for rougthly the same reasons as above. More specificially, motifs can occur inside these domains in regions that are otherwise disordered, but are too short to split the homologous match in two. You can, however, turn on this filter (by clicking 'Yes').
Motif over-representation settings
Our analysis of known linear motifs, within interactome datasets, shows that they typically occur in a fraction of the total sequences quried. This has to do with many things: interactions might be false-positives or proteins might interact in different ways - that is, a hub protein can have multiple binding sites. There are several parameters that you can tune to find motifs:Min # of sequences with motif: this is the minimum number of sequences (in each set) that are required to have any motif; i.e. if you set this to 5, it means that at least 5 sequences must contain at least one instance of a motif in order for that motif to be kept.
Min # of fixed (non wildcard) residues in motifs: this is the number of motif positions required to be fixed, that is that are not 'x' or '.'. Note that if you specify groups (see below) that these then count as fixed positions.
Max motif length: this is the maximum length (stretch of sequence) for any discovered motif. The default value (8) is typical of most motifs, though some motifs as long as 15 residues are known. Generally, for longer motifs, shorter (i.e. within 8 residues) are also found.
Max number of motifs to keep (per hub): this is the maximum number of motifs to be kept for display.
Max set size (interactions only, per hub): very large sets of proteins can give rise to funny results, and can moreover take a long time. We generally dont' consider sets larger than 100, but you can raise this value if you are really interested.
Amino acid groups to be used (max 6): There are several ways to group amino acids when defining motifs. This allows you to do this. The default groups (DE KR ST ILMVF) are those we defined based on an analysis of known linear motifs, though there are other things you might wish to try (e.g. FYW for aromatic, STPGA for small, etc.). Note that you can't, currently, have amino acids in more than one group. Note additionally that the method will always run first without grouping, meaning that you can find both PxxPxR and PxxPx[KR] in the same run.
Group motifs in output (blank means do not group): (Experimental) this allows you to group motifs by their similarity in terms of where they lie in the sequences. The parameter ranges from 0-1 and is a measure of how similar the instances are for a pair of motifs (Jaccard score). This can be useful if one is looking for discrete kinds of motifs in the same set of sequences. Try a value of 0.3 or 0.5. Note that this currently means the motifs that are grouped only have one (the best) representative kept.
Other parameters
Surface binding (PepSite) / Min % sequence identity for structure: The best results for PepSite come from exact structures (i.e. of the protein hub of interest) or very close homologues (e.g. >= 70% identity; meaning human/mouse or similar). Lowering this can be tried, though be warned that PepSite doesn't do anything to model the structures, so that you migth get a significant hit on the surface of a protein that is very different from your hub, say, if you set this to 30% or less.Summary table
Once the job has run, you will be presented with a table, something like this: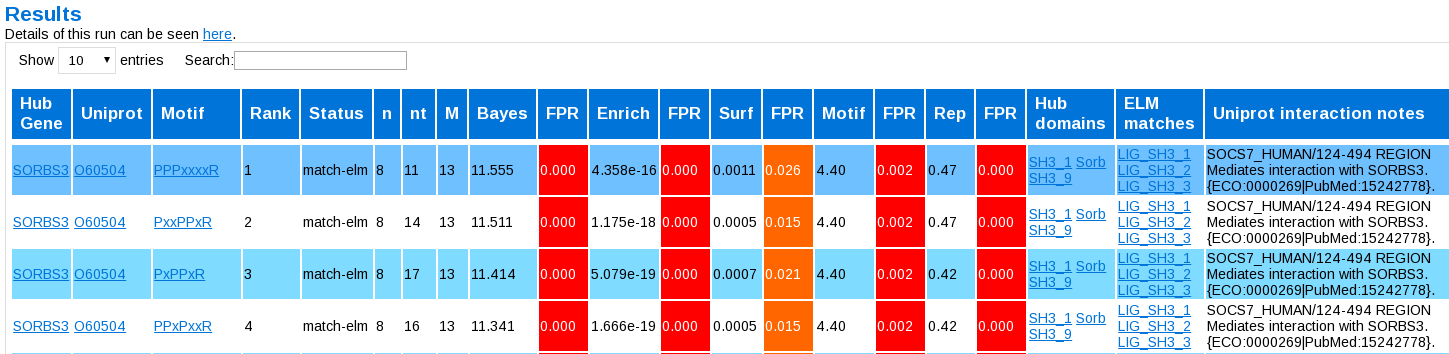
The columns are:
- Hub Gene: the name of the hub (if given)
- Uniprot: a link to the Uniprot entry (if available)
- Rank: the original position of the motif in the list
- Status: novel or a match to an already known motif
- n: the number of proteins in the set that have at least instance copy of this motif
- nt: the number of instances of the motif in the set (i.e. where proteins can have more than one)
- M: the total number of proteins used in the set
- Bayes: the value of the Bayesian combination metrix
- FPR: the false positive rate associated with this value
- Enrich: the corrected P-value for the motif over-representation or enrichment
- FPR: the false positive rate associated with this value
- Surf: the PepSite p-value for how well the best instance fits on the best protein structure (if any)
- FPR: the false positive rate associated with this value
- Motif: how 'motif-like' this motif is in terms of the amino acids inside it
- FPR: the false positive rate associated with this value
- Rep: how well this motif overlap with other significant motifs
- FPR: the false positive rate associated with this value
- Hub domains: Pfam-A domains found in the Hub protein (if given)
- Elm matches: Instances where the ELM motif is found interacting with the right domain/protein hub (if any)
- Uniprot interaction notes: Uniprot annotations linking motif instances to the hub protein (if any)
Hub pages
Pages for each hub are linked from the above table. When you first click on them, you are presented with something like this:
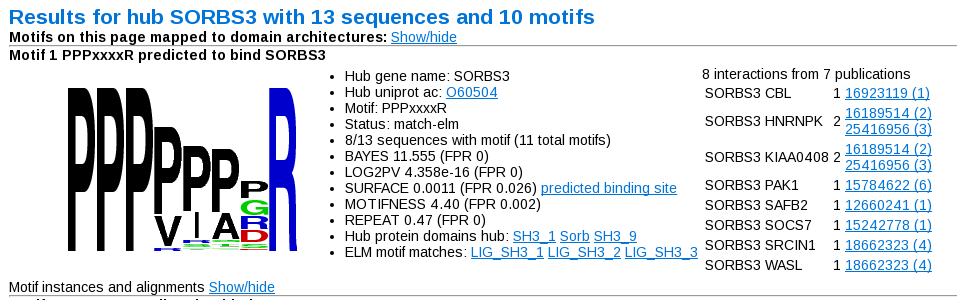
You first see a sequence logo, derived based on all the instances found in the sequences. Next to this is a summary of the results from the previous table page. On the right is a summary of the interactions used to define this particular motif. The number next to the interacting pair is the number of sources (i.e. labels) in which this particular interactino was seen. For each of the sources (or publications), listed on the right of this table, the number gives the total number of interactions from this source (i.e. whether the source is high- or low- throughput). Strings of numbers given in the sources (separated by spaces or punctuation) are presumed to be pubmed identifiers, and hyperlinked accordingly.
At the top of the page you can show the location of the motifs on domain diagrams, to get something like this:
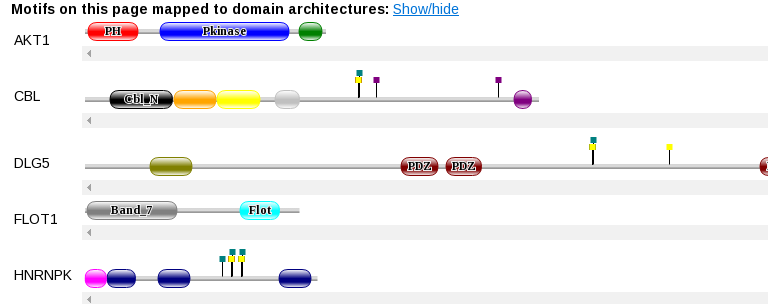
Which should be self-explanatory.
For each motif you can also see more detailed information about each instance of the motif by clicking the "Show/hide" toggle next to "Motif instances and alignment". You will then get someting like this:
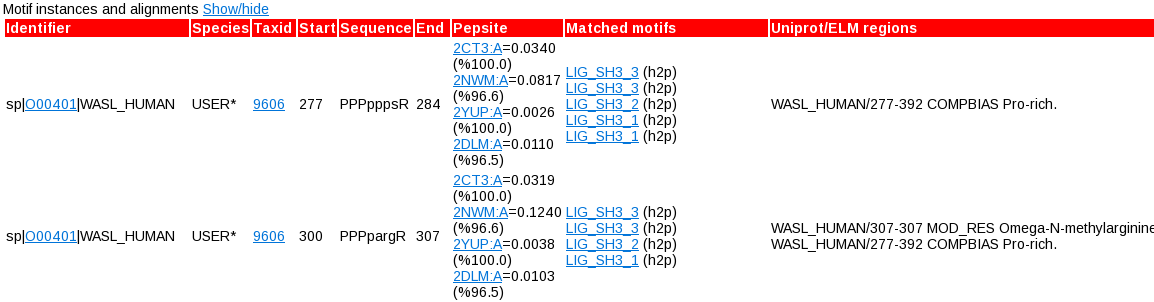
Here there are numerous hyperlinks to Uniprot, NCBI taxonomy (in the case of orthologues), PepSite links for each structure considered, and protein domains. Here you will also see additional overlaps to ELM motifs (ie. all of them - virtually every stretch of sequence overlaps with some motifs as they are very degenerate), and additional Uniprot regions that overlap with each motif (i.e. regardless of whether they mention the Hub protein or not). Here you can see things like post-translational modifications, effects on other proteins, disease variants, etc. that can help you decide whether or not a motif is interesting.
Practical hints
- In general, you will have a much better time with this tool if you use Uniprot accessions, identifiers or gene-names, as opposed to sequences that you find. The underlying database is very Uniprot centric, meaning that a lot of links are available if you adopt this strategy that are missed if you just paste in sequences from some other source.
- It is usually helpful to spend some time tuning your interactions to your interest. Just taking a long list of noisy interactors for a hub of interest will work, in the sense that the tool will work, but noise, in the form of dubious interactors, might mean that real motifs are swamped (and thus missed). If you know that an interactor interacts with your hub in a different way (i.e. not via a motif, but via some large protein-surface interactions), then it is better possibly to leave it out.
- Default filtering is optimised for ELM motifs, but might not be optimal for your set of sequences. It is worth trying different combinations of filters to see what comes out.
- Equally for the wild-cards used. You can experiment with this too, particularly if you have some ideas (e.g. phosphorylation) already about what residues might be important for the interactions
- For large sets, you should consider raising the "Min # of sequences with motif:" - e.g. if you have 50 sequences, consider raising it to 10 or higher. Large sets will always have motifs with low counts (e.g. < 10), which naturally the statistics should cope with, but this doesn't always work, particularly as real motifs are often on the border of significance